Dispositifs médicaux
Audit croisé "évaluation du management de la qualité du circuit des DMI dans les établissements de santé"
Les OMéDIT Bretagne et OMéDIT Normandie lancent une campagne d’audits inter-structures ouverte à l'ensemble des établissements de santé bretons et normands, avec une activité de chirurgie.
Cet audit a notamment pour objectifs de/d' :
- Susciter un dialogue interprofessionnels et inter-établissements sur la mise en œuvre du système de management de la qualité des DMI
- Comparer les pratiques professionnelles de santé à partir d’une méthode et d’un référentiel commun
- Évaluer l’organisation mise en place pour garantir la sécurisation du circuit des DMI
- Évaluer la complétude des informations de traçabilité sanitaire
- Identifier les points forts et les axes d’amélioration
- Élaborer un plan d’actions
Vous êtes professionnel de santé d’un établissement breton ou normand avec une activité de chirurgie ?
⇒ Un webinaire de présentation de la campagne 2024-2025 est prévue le Jeudi 19 Septembre à 16h

Adaptation des dispositions transitoires relatives à certains DM
Le règlement (UE) 2023/607 du Parlement européen et du Conseil du 15 mars 2023, applicable depuis le 20 mars 2023, modifie les délais de mise en conformité pour :
- Les dispositifs dont l’intervention d’un organisme notifié n’était pas nécessaire sous les directives mais qui s’impose sous les règlements pourront être mis sur le marché jusqu’au 31 décembre 2028
- Les dispositifs de classe I stérile, I sur mesure, Iia, IIb non implantables jusqu’au 31 décembre 2028
- Les dispositifs IIB implantables et III jusqu’au 31 décembre 2027
- Les dispositifs de classe III sur mesure jusqu’au 31 décembre 2026
L’application de la période de transition prolongée est soumise à plusieurs conditions cumulatives :
- Les dispositifs restent conformes aux directives
- Les dispositifs ne subissent pas de modifications significatives de leur conception et destination
- Les dispositifs ne présentent pas de risque inacceptable pour la santé ou la sécurité des patients
- Au plus tard le 26 mai 2024, le fabricant a mis en place un système de gestion de la qualité
- Au plus tard le 26 mai 2024, le fabricant ou son mandataire a introduit une demande de certification auprès d’un organisme notifié
Enfin, le délai de vente limite du 27 mai 2025 (date au terme de laquelle les DM ne pouvaient plus être mis à disposition sur le marché de l’UE) est supprimé sans condition
L'article 27 de la loi DDADUE, applicable depuis le 10 mars 2023, a adapté le CSP aux règlements européens pour les DM afin que :
- la suspension ou l’arrêt de leur commercialisation soient notifiés à l’ANSM par le fabricant, son mandataire, l’importateur et le distributeur hors détail qui prennent la décision ou sont informés de faits susceptibles d’entraîner une telle interruption ;
- les opérateurs précités ayant identifié un risque dans la prise en charge de l’état de santé du patient mettent en œuvre toute mesure utile et nécessaire par anticipation pour éviter la rupture et assurer la continuité de la prise en charge ;
- ces opérateurs déclarent à l’ANSM tout risque de rupture ou toute rupture ;
- l’ANSM mette en œuvre toute mesure utile et nécessaire pour éviter la rupture et assurer la continuité de la prise en charge, pour tout risque de rupture ou rupture constatés par l’ANSM n’ayant pas fait l’objet des mesures ou déclaration précédemment décrites, après consultation des opérateurs précités, des professionnels de santé et des associations de patients et d’usagers.
Entrée en application du nouveau règlement européen relatif aux dispositifs médicaux
Le règlement européen UE 2017/745 relatif aux dispositifs médicaux est entré en application le 26 mai 2021. Il vise à renforcer la sécurité des dispositifs médicaux et introduit de nouvelles exigences :
L'identifiant unique du dispositif (IUD) :
L'IUD est un système d'identification reposant sur :
- l'attribution d'un IUD à tous les dispositifs (exceptés DM sur mesure et DM d'investigations cliniques)
- l'apposition de l'IUD sur l'étiquette ou sur le DM et sur tous les niveaux de conditionnements supérieurs (sauf conteneurs d'expédition)
- l'enregistrement dans la base de données européenne des DM (EUDAMED)
- l'enregistrement dans les sytèmes informatiques des opérateurs (fabricant, distributeur, mandataire, importateur, établissement)
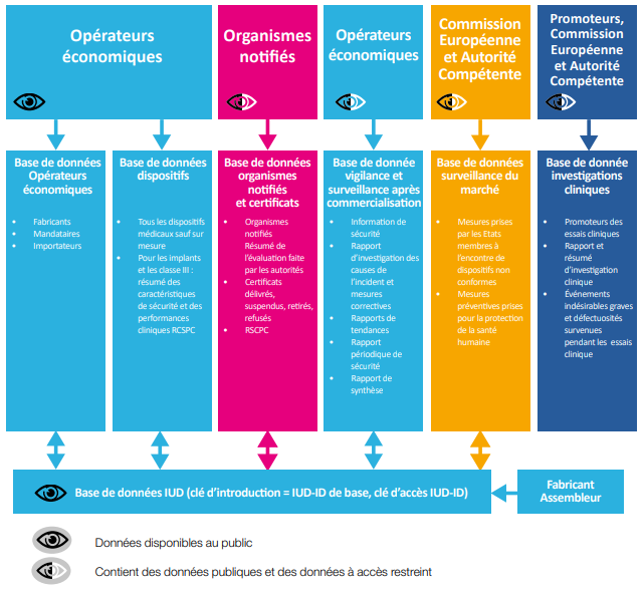
La base de données européenne des DM : EUDAMED
La base de données EUDAMED, composée de 6 modules, va permettre
- d'avoir accès à des informations sur les DM (résumé des caractéristiques de sécurité et des performances cliniques, classe de risque du dispositif, mises en garde ou contre-indications importantes, ...)
- de connaître les incidents déclarés et l'avancée des investigations cliniques
- de renforcer la coordination et la coopération entre les différents acteurs
La carte d'implant et informations à fournir au patient avec un dispositif implantable :
Les fabricants et les établissements doivent fournir des informations au patient (identification du dispositif, mises en garde à prendre par le patient ou par un professionnel de santé vis à vis des examens médicaux, durée de vie du dispositif, ...) ainsi qu'une carte d'implant remise avec le DMI (sauf certains DMI)
Le Medical Device Coordination Group a publié un guide sur les informations à fournir aux patients avec des exemples de présentation pour la carte d'implant (cliquez ici).
Liens utiles
- Jeudis de l'IUD (Europharmat et SNITEM)
- Réunion d’information sur le nouveau règlement européen relatif aux DM
- L’IUD et de son impact dans le circuit des dispositifs médicaux dans les établissements de santé (Europharmat et SNITEM en collaboration avec la DGOS)
- Guide sur l'application du règlement européen relatif aux dispositifs médicaux à destination des établissements de santé (Europharmat et SNITEM, 2024)
Arrêté du 8 septembre 2021 relatif au management de la qualité du circuit des dispositifs médicaux implantables dans les établissements de santé et les installations de chirurgie
Arrêté relatif au système de management de la qualité du circuit des DMI dans les établissements de santé et les installations de chirurgie esthétique
- Mettre en oeuvre le management de la qualité du circuit des DMI dans les établissements de santé et les installations de chirurgies esthétiques
- Sécuriser et améliorer l'organisation du circuit du DMI à toutes les étapes du circuit
- Améliorer la traçabilité sanitaire des DMI
Liens utiles :
- Organiser le circuit des DMI (ANAP, Octobre 2023)
- Volet Traçabilité des DMI (ANS)
Arrêté du 22 décembre 2021 subordonnant la prise en charge des produits de santé inscrits sur la liste prévue à l'article L. 165-11 du code de la sécurité sociale au recueil et à la transmission de certaines informations relatives à leur usage
Liste des produits de santé financés au titre des prestations d’hospitalisation prévue à l’article L. 165-11 du code de la sécurité sociale dite "intra-GHS"
L'arrêté du 22 décembre 2021 et la note d'information interministérielle du 22 décembre 2021 précisent les modalités de recueil et de transmissions de certaines informations par les établissements en vue d'améliorer le suivi et d'analyser l'usage en vie réelle des dispositifs médicaux inscrits sur la liste "intra-GHS".
La liste consolidée des DM "intra-GHS" est disponible sur le site du ministère des solidarités et de la santé. Les DM inscrits sur cette liste appartiennent à des catégories homogènes de dispositifs et doivent faire l'objet au préalable d'une évaluation par la commission nationale d'évaluation des dispositifs médicaux et des technologies de santé de la HAS.
Le recueil et la transmission des informations au moment de la facturation ou de la valorisation des prestations d'hospitalisation concernent :
- le numéro FINESS de l'établissement
- le numéro administratif de séjour
- la date d'utilisation
- le code IUD-ID du DM
- le nombre d'unités utilisées
Les informations recueillies sont transmises à l'ATIH sous la forme d'un fichier DATEXP dans le cadre du suivi de l'utilisation à travers le programme de médicalisation des systèmes d'information (PMSI).
La mise en oeuvre de ce recueil est possible à compter du 1er janvier 2022 pour les établissements volontaires et sera obligatoire à compter du 1er avril 2022 pour tous les établissements de santé (ex-DG et ex-OQN).
Boite à outils OMéDIT/ResOMéDIT
L'OMéDIT Normandie a réalisé un webinaire sur le suivi de l'utilisation des DM intra-GHS et IUD : vidéo du webinaire et support de présentation
L’OMEDIT Nouvelle-Aquitaine Guadeloupe a élaboré un support de communication à destination des patients : information transmise à la suite d’une implantation chirurgicale de Dispositifs Médicaux Implantables
- Flyer à destination des patients "Vous êtes porteur d'un implant"
- Film : DISPOSITIFS MÉDICAUX IMPLANTABLES : TOUS ACTEURS !
L'OMéDIT Centre Val de Loire a élaboré
- un flyer récapitulatif sur la mise en oeuvre du suivi des DM de la liste intra-GHS
- une trame type du bilan annuel du responsable du système de management de la qualité
Le ResOMéDIT met également à disposition des outils pour accompagner les établissements dans le déploiement de l'IUD et l'informatisation des DMI :
Les OMéDIT du Grand Ouest (Bretagne, Centre - Val de Loire, Normandie et Pays de la Loire) propose une fiche de poste du responsable du système de management qualité du circuit des DMI
L'OMéDIT Grand Est met à disposition des fiches informatives à destination des patients sous forme de triptyque
Cartographie de l'informatisation des DMI :
L'OMéDIT propose un accompagnement aux établissements de santé de la région pour réaliser une cartographie de l'informatisation du circuit des DMI conçue par le ResOMéDIT
Les objectifs :
- Recenser les logiciels et la nature des données informatisées dont l'IUD
- Réperer les niveaux d'interopérabilité des logiciels
- Analyser le niveau d'informatisation du circuit du DMI
- Etablir un plan d'action pour optimiser les données du circuit des DMI
L'accompagnement :
- Présentation de la cartographie lors d'un webinaire en septembre-octobre
- Echanger sur les difficultés rencontrées pour l'informatisation du circuit des DMI à partir de la cartographie
- Partager des actions opérationnelles sous forme de retours d'expérience régionaux/nationaux
Traçabilité des DMI
Matériovigilance :
- Petit guide du parfait matériovigilant : affiche
- Petit guide du parfait matériovigilant : format de poche
Outil d'auto-évaluation performance du circuit des Dispositifs Médicaux Stériles (DMS) et des Dispositifs Médicaux Implantables (DMI) :
Traçabilité sanitaire des DMI :
- Décret n°2026-299 du 17 avril 2026 relatif aux dispositifs médicaux (NOUVEAU)
- Le guide de traçabilité sanitaire des DMI proposé par Europharmat (2025)
- Le guide de poche "Traçabilité des DMI" proposé par l'OMéDIT Normandie
- Quels sont les dispositifs implantables soumis à traçabilité obligatoire ? Fiche de bonnes pratiques proposée par l'OMéDIT Centre
- Note d'information DGOS/PF2/2019/69 du 27 mars 2019 relative à la traçabilité des DMI dans les établissements de santé et aux outils d’autoévaluation et d’accompagnement disponibles
- Instruction DGOS/PF2 n° 2015-200 du 15 juin 2015 relative aux résultats de l’enquête nationale sur l’organisation de la traçabilité sanitaire des DMI dans les établissements de santé des secteurs publics et privés, titulaires d’activités de médecine, chirurgie et obstétrique